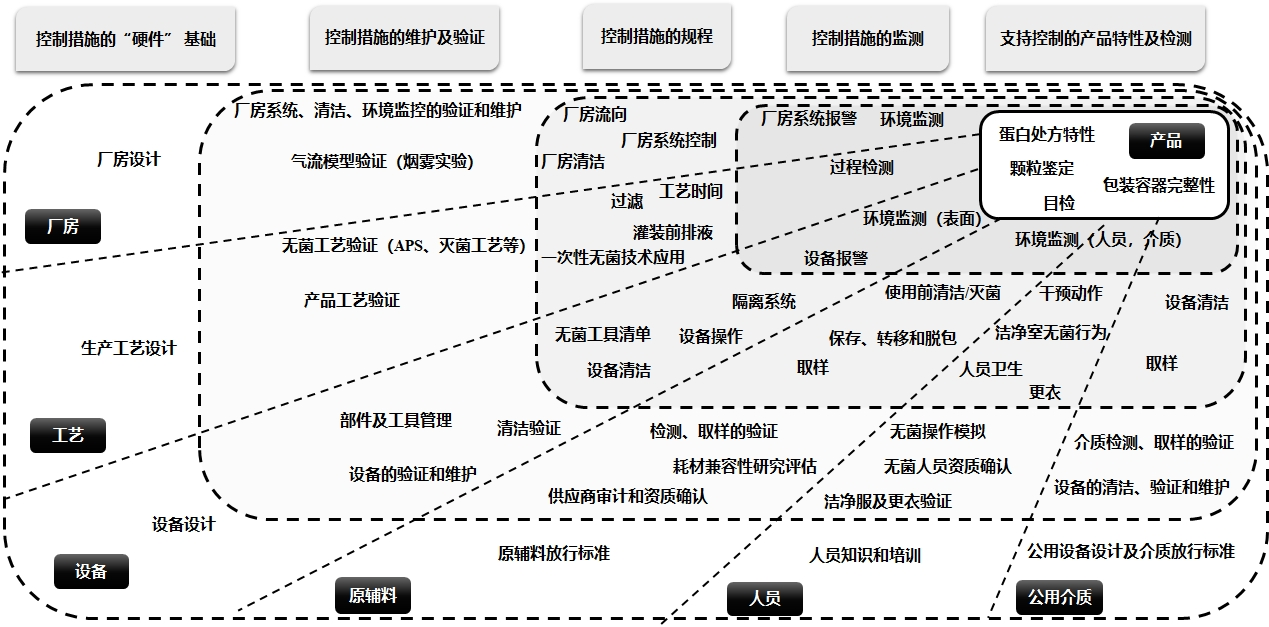
ECA 注射剂目检指南与 USP 1790 注射剂目检对比解读
ECA 注射剂目检指南与 USP 1790 注射剂目检对比解读
作者:识林向导@沐清风,审校:识林向导@Xunger
ECA 注射用药品的目检4.0版本于2024.3发布,该指南视为对不同药典个论的额外补充,本文以ECA 注射用药品的目检4.0版本为基础,对比解读USP1790中的相关内容,探讨注射用药品100%目检的行业最佳实践,英文部分为指南原文,ECA中文部分来自识林翻译审校版,对比解读部分为作者的思考和解读,供参考。
目录[隐藏] |
人工目检
| 要求 | ECA | USP 1790 |
| Illumination 照度 | Illumination: The intensity of the illumination at the inspection point should have at least 2000 lux. For Blow-Fill-Seal Products an illumination of 10.000 lux is recommended. The accuracy of the lux-meter should be considered. The color reproduction, using the CRI index, should preferably have an RA value > 90%. Illumination should be regularly qualified and part of the preventive maintenance programme. An appropriate interval for checking the illumination is 6 months. It is recommended that the technical measurement used to determine the light intensity is performed at a prefixed point, which should be very close to the inspection point of the operator. The ambient illumination must not interfere with the illumination of the workplace and should be turned down during inspection, if possible. Reflecting surfaces should be avoided. | 6.1 Manual Visual Inspection Chapter 790 recommends that the minimum light levels be not less than 2000–3750 lux at the point of inspection for routine inspection of clear glass containers. This range was chosen to harmonize with current European Pharmacopoeia requirements (59) and is further supported by recent studies performed in Japan (61) as part of a review of the current Japanese Pharmacopoeia requirements (60). Special attention should be given to ensure that inspection is not performed below the lower limit of 2000 lux. Increased light levels are recommended for translucent plastic containers or those made from amber glass. Under these circumstances, light levels as high as 10,000 lux may prove beneficial. The frequency of monitoring should be based on historical experience with the type of light source in use. A lower light-intensity action limit should be established to trigger corrective action before inspection is performed below the lower limit of the range. |
| 对比解读: 1、检测点的照度强度至少应为2000 lux,USP 1790 要求为不低于2000–3750 lux; 2、对于吹灌封产品,ECA建议照明度为 10000 lux;对于半透明塑料包装或琥珀色玻璃包装应考虑增加照度,某些特殊情况下,照度提高到10000lux。 3、ECA建议应该考虑照度计的准确度,使用 CRI 指数的色彩再现最好具有 RA 值 > 90%。 (注:a. JB/T 20186-2017可见异物灯检仪中要求,设备4.2.2灯检仪的光源均匀性偏差应不大于6%,灯检仪的光源稳定性偏差应不大于4%;b. 实际执行建议结合照度计准确度,照度设置范围适当缩窄留出余量) 4、 ECA建议的检查照明的适当间隔建议为 6 个月。 延伸补充观点: 1、中国药典:用无色透明容器包装的无色供试品溶液,检查时被观察供试品所在处的光照度应为1000~1500lx;用透明塑料容器包装、棕色透明容器包装的供试品或有色供试品溶液,光照度应为2000~3000lx;混悬型供试品或乳状液,光照度应增加至约4000lx;
2、不同照度对产品质量影响,一般可以通过产品光照影响因素稳定性实验进行评估,根据《中国药典2020 四部 9001 原料药物与制剂稳定性试验指导原则》,光照影响因素稳定性实验条件一般照度为4500lx±500lx,详见识林主题词:光稳定性; | ||
| Ambient conditions 环境 | Ambient conditions: Ambient conditions have a high impact on the quality of this operation. Temperature should be room temperature and not exceed 25 °C in the summer if not otherwise justified. The relative humidity and air velocity should be controlled and ensure comfortable working conditions. The noise level should therefore be below 55dB. | The inspection room environment should also be considered. Temperature and humidity should be controlled for inspector comfort. Reduced ambient lighting is recommended to focus the inspection process and to reduce distraction from extraneous reflections. Special care should be given to inspection rooms with exterior windows that allow daylight into the room, thereby changing ambient lighting throughout the day and with the seasons. |
| 对比解读: 1、ECA对于目检环境的描述较USP1790更加具体,温度应为室温,如无其他理由,夏季不得超过 25°C。应控制相对湿度和空气速度,确保舒适的工作条件,噪音水平应低于55dB; 2、USP 1790建议降低室内灯光的亮度,来增加人员目检程序的专注度,降低外界反射对于人员检测的影响,尤其要注意安装窗户的目检房间,外界光可进入目检房间,随着时间或季节的交替,室内的光线会变化。 延伸补充观点: 中国药典:中国药典2020 四部 0904 可见异物检查法,第一法(灯检法)灯检法应在暗室中进行(注:暗室一般意义上理解为无窗户,室内照明光源关闭)。 | ||
| Personal 人员 | Personnel involved in the visual inspection should undergo regularly eye tests. The optometrist should focus on the ability to discriminate small differences in uniform structures, e.g. open/closed circles. Also, color vision of the inspector should be tested. 参与视力检查的人员应定期接受视力检查。验光师应注重辨别均匀结构中的细微差别的能力,例如开放/封闭的环形。此外,还应测试检查员的色觉。There is sometimes the expectation for operators to wear corrective lenses when undergoing visual inspection qualification, if they are worn during daily life.如果操作员在日常生活中佩戴矫正镜片,那么在进行视力检查鉴定时,有时会期望他们佩戴矫正镜片。The requested medical check examines e.g. three-dimensional sight, color identification, stigmatic corrections and most importantly reflecting the normal "daily life" visual capability. This ensures that the operator can see defects. 所要求的体检包括检查三维视觉、颜色识别、散光矫正、以及最重要的“日常生活”正常视力。这确保操作员能够看到缺陷。 The medical check is not related to the competence of performing visual inspection activity.体检与执行目检活动的能力无关。 This is checked during the qualification of the operator and whether he has to wear his glasses is determined in this process. 这些检查是在操作员资质确认过程中进行的,由此确定操作员是否必须戴眼镜进行目检。 | Before training, potential inspectors should be tested for visual acuity (69) and color perception. Near-vision performance should be the equivalent of 6/6 m (20/20 ft), but not conducted at this distance, and with no impairment of color vision. Both the Snellen and Jaeger charts are useful for verifying visual acuity; they test far and near vision, respectively. The use of corrective lenses to achieve the desired visual acuity is permitted. |
| 对比解读: 1、ECA和USP 1790均规定了注射剂目检人员应该接受视力和色觉的定期检查,其中USP 1790提到了视力检查应包括远视力和近视力检查。 2、ECA和USP 1790均规定了操作员可以戴眼镜进行目检,ECA给出了是否需要佩戴的测试检查方法。 延伸补充观点: 中国药典:按照药典要求,人员远距离和近距离视力测验,均应为4.9及以上(矫正视力应为5.0及以上),应无色盲,还应注意人员从相同形状事物中区分微小差别的能力。(注:实践中需注意,药典采用的是标准对数视力表按照五分记录法,5.0即为正常标准视力值,如果采用 “国际标准视力表”小数记录法需进行换算)。 | ||
| Training and qualification 培训和资质考核 | Personnel performing visual inspection must be qualified, comprising initial qualification and periodic requalification. Initial qualification should follow a predefined schedule, starting with the introduction of the new employees to training kits. These training kits should contain all kind of defects and should be updated constantly with new evolving defects out of production. These training kits should be specific for the dosage form. In case of unstable defects or defects which are not available photos of defects or artificial defects may be used. 执行目检的人员必须经过资格审查,包括初次资格审查和定期重新资格审查。初始资格认证应遵循预先定义的时间表,从向新员工介绍培训套件开始。这些培训工具包应该包含所有类型的缺陷,并且应该随着生产中出现的新缺陷而不断更新。这些培训套件应该针对特定的剂型。对于不稳定的缺陷或无法获得的缺陷,可以使用缺陷照片或人工缺陷。 A bracketing approach for similar products is acceptable. Following the training via training sets there should be a side to side training with an experienced operator. The trainee should have the chance to ask questions with the experienced operator performing a parallel 100 % inspection of the inspected units of the trainee. 对于类似产品采用括号法是可以接受的。通过训练集接受培训之后,还应接受与熟练目检员并行操作的培训。在对被检单元进行并行100%目检时,受训人员应有机会向熟练目检员提问。 Following successful completion of the side by side training the initial qualification should be performed with a qualification set. This set should focus on critical and major defects and also contain a limited number of minor defects. The number of units to be inspected should represent the number of objects which are inspected in the routine process from one eye break to the next eye break. 成功完成并行培训后,应使用确认集进行初始资质确认。该确认集应着重于关键缺陷和主要缺陷,同时也包含有限数量的次要缺陷。被检单元数应代表常规目检过程中相邻两次休息之间接受目检的物品的数量。 There is sometimes the expectation for operators to be qualified taking into account different stress factors, including fatigue at the end of shift. It can be doubted that a fatigue effect can be simulated in an examination situation with the impact for the operator, whether he/she can keep his/her job. With this there is always nervousness and some people have even examination anxiety which leads to release of Adrenalin in the human body, which counters a fatigue effect. Additionally, when performing initial qualification, the fatigue at the end of shift can't be simulated anyway, as the operators are not yet allowed to perform a "normal" visual inspection workday. 有时会期望操作员资质确认考虑不同的压力因素,包括班次结束时的疲劳。值得怀疑的是,在考核情境(决定操作员能否保住工作)中是否能够模拟疲劳效应。在考核情境下,人们总是会感到紧张,有些人甚至会有考试焦虑,这会导致人体释放肾上腺素,以抵消疲劳效应。此外,在进行初始资质确认时,无法模拟班次结束时的疲劳,因为操作员此时还不被允许执行“正常”的目检工作。 A set of product specific containers with real product, with 10-20% but less than 30% of the containers containing randomly distributed defects. The sets should be cleaned after usage and routinely checked for defects at least every 6 months. There should be a logbook for each test set. 一组装有真实产品的容器,其中10-20%(但少于30%)的容器有随机分布的缺陷。该测试集应包含所有已知缺陷,使用后应清洁测试套件,并至少每6个月定期检查缺陷。每个测试集都应设立日志。 | Training should include a phased approach with a specified number of training hours expected for each segment. Initially, train the potential inspectors with defect photographs or a video library and clear, written descriptions. Utilize subject matter experts to mentor and provide hands-on training with defect standards for the specified method. Utilize subject matter experts to mentor and provide hands-on training with defect standards for the specified method. Reinforce mental or silent counting and follow the paced sequence to achieve consistent inspection timing. Qualification should be performed for each product type and package that the inspector will encounter. A bracketed or matrix approach can be used to simplify qualification of products with similar physical or visual characteristics such as container type and size, dosage form, product viscosity, color, and others (product families or groups). It is common to initially train and qualify personnel on clear solutions in clear containers (if produced at the facility) and then expand their expertise to the inspection of more difficult dosage forms or presentations. Acceptance criteria should reflect the different risk categories (i.e., critical, major, and minor) of these defects when combined into a single test set, and a single acceptance criterion should not be used with a set with multiple defect risk categories. Inspector fatigue may be addressed in the qualification process by testing under worst-case conditions (e.g., at the end of a typical inspection shift). 7.5 Test Sets The number of defective units in each test set should be limited to approximately 10% to prevent rejection bias (62). |
| 对比解读: 1、ECA和USP 1790 对于目检人员的培训均要求通过分阶段的方式,先从了解缺陷品开始,然后由有经验的分析员手把手的现场教学和平行测试,再到使用确认集进行资质确认。 2、ECA和USP 1790中均规定了对于类似的产品的培训和考核可以采用括号法。 3、ECA规定了确认集(目检人员资质确认标准品)的数量应该能够代表日常目检时每次检测的数量。 4、ECA要求确认集的缺陷品占比约10-20%(但少于30%),USP 1790要求确认集的缺陷品占比约10%。 5、ECA和USP 1790均提到了对于操作员资质确认应该考虑不同的压力因素,应基于最差条件,例如:在完成一个班次的目检后; ECA指南对于此要求有不同的解读,认为人员在考核情境下,人们总是会感到紧张,有些人甚至会有考试焦虑,这会导致人体释放肾上腺素,以抵消疲劳效应,并且在进行初始资质确认时,无法模拟班次结束时的疲劳,因为操作员此时还不被允许执行“正常”的目检工作。 延伸补充观点: 1、ECA Visual Inspection Group 《Questions & Answers Document Version 2.0》:MI4 Q:Should the set of samples for the qualification (training) of the staff be taken directly form the production rejects or should it be produced artificially based on typical defects?
2、FDA 行业指南:注射产品可见异物检查:For personnel qualification and automated inspection systems validation, a mixture of good injectable product units and defective units containing visible particulates should be used (Melchore 2011). This test set should be prepared and approved by quality assurance staff. Manufacturers should develop libraries of defective units from samples collected throughout the product life cycle, samples created to simulate production defects, or samples purchased to be representative of the types of particulates likely to occur for the drug product and itsmanufacturing process. Quality assurance staff should review the library of defective samples and compare the samples to established standards for proper classification. The library should contain examples from the lower limits of visual detection determined in the threshold studies. If a new particulate matter defect is identified, it should be analyzed to determine its source and added to the training library. 对于人员资质确认和自动设备验证的,应使用合格的产品和标准缺陷品的组合。该“确认集”(有的叫测试集)由QA准备和批准。从产品的生命周期中收集缺陷样品,委外制备的缺陷样品或者药品及其生产过程中可能产生的异物类型而购买的样品中建立缺陷样品库。QA应检查缺陷样品库,并将样品与批准的标准进行比较,从而对缺陷样品进行分级。该缺陷库中应包含检测阀值研究中确定的视觉检查下限的缺陷样品。如果在生产过程中发现了新的缺陷样品,应该对该缺陷进行分析并确认其来源,并将该类型的缺陷加入到“培训集”中。 3、关于人工确认集10%的缺陷率的科学依据补充:Studies by Wolfe et al (2007) and Rich et al (2008) indicate that the probability of detection during human visual inspection is influenced by the defect rate. PoD increases with increasing defect rate. Ideally test sets would be prepared with a defect rate equivalent to that observed in routine production inspection — typically 1–2% (Shabushnig, 2014). It is not uncommon to require 100 defect examples which would result in test sets of 10,000–20,000 in size. Such large sets are not practical, especially when multiple test sets are often required to represent the range of products and containers manufactured at a given location. As such, a practical compromise can be made by not exceeding a defect rate of 10% resulting in test sets of approximately 1,000 when 100 defect examples are included. This limits the degree of bias and better reflects true performance in routine inspection. 研究表明,人类视觉检查期间的检测概率受到缺陷率的影响。PoD随着缺陷率的增加而增加。理想情况下,准备的测试集的缺陷率与常规生产检验中观察到的缺陷率相当—通常为1–2%(Shabushnig, 2014)。需要100个缺陷示例并不罕见,这将导致测试集的大小为10,000–20,000。这样大的测试集是不实际的,尤其是当经常需要多个测试集来代表在给定位置生产的产品和容器的范围时。因此,当包含100个缺陷示例时,可以通过不超过10%的缺陷率得到大约1000个测试集来进行实际折衷。这限制了偏差的程度,并更好地反映了日常检查的真实性能。 | ||
| Acceptance criteria for the qualification should be predefined. In the initial qualification all critical defects and a predefined level for major defects should be found. Also, a limit for the False Rejects (rejection of samples without defects) should be predefined (e.g. 5-10%). 应该预先定义资格的验收标准。在初始鉴定中,应发现所有严重缺陷以及主要缺陷的预定义级别。此外,应该预先定义错误拒绝(拒绝没有缺陷的样品)的极限(例如 5-10%)。 Routine requalification must be performed at least every 12 months. After a second failure the operator must undergo a repeated eye test and a subsequent new qualification with the training kit and a subsequent new qualification via a qualification kit. 必须至少每 12 个月进行一次例行重新鉴定。第二次失败后,操作员必须再次接受视力测试,然后使用培训套件进行新的资格认证,最后通过资格认证套件进行新的资格认证。 | Acceptance criteria for each defect class should be based on the PoD, or RZE observed during test set qualification for the package. PERCENT RECOVERY QUALIFICATION METHOD: A test set is prepared with bracketing of each particle type with a one-seeded unit being near-threshold (just above 70% PoD) and the other larger with a higher PoD. Typically, not less than 80% or 90% of the units containing visible particles are expected to be detected from the qualification test set. RZE QUALIFICATION METHOD: The previously determined test set RZE is used to assess the results obtained by the inspector to be qualified. Each inspector to be qualified would then inspect the blinded set, and the RZE (the average PoD for all units containing particles >70% PoD) is calculated for each trial. The acceptance criterion is to achieve an RZE during the qualification that is equal to or better than the RZE originally determined during the threshold study. %PoD = (Number of times rejected)/(Number of times inspected) × 100 RZE = the average of all PoD values for all units in the test set with an individual %PoD >70% When assessing inspector performance, a limit is also needed for false rejection. A target of Inspectors should be requalified at least annually. Requalification includes a test of visual acuity and testing with at least one product/test set configuration. A single successful inspection of the test set is sufficient for requalification. Requalification may also be necessary if poor performance is observed during routine inspection or if the inspector has been away from the inspection operation for an extended period (e.g., more than 3 months). If an inspector fails the requalification test, a retraining process should be initiated to identify the root cause and allow the inspector to receive additional instruction. After this process has been completed, the inspector may attempt to meet the acceptance criteria one additional time. If the inspector fails, he or she may attempt to qualify again after a specified time period. | |
| 对比解读: 1、对于人员确认的方法和标准,1790列举了两种,POD或RZE,并列出了详细的计算公式。 2、ECA和1790均要求考察误检率,1790建议的误检率低于5%,ECA建议5-10%。 3、ECA和1790均规定人员定期再确认的周期至少为每年;ECA规定了人员资质确认第二次测试失败后需要重新进行视力检查和测试集考核,1790规定每次的再确认均需要视力测试和测试集考核。 4、1790规定了除定期再确认外,当日常发现人员检测表现差或人员脱离检测较长(例如3个月以上)时也需要考虑再次确认。 5、1790规定了人员再确认至少选择一个产品,至少进行一次成功的测试集考核。 延伸补充观点: EU GMP Annex 1:8.31人工检查时,应在有适当照明和环境的受控条件下进行。检查频率应经适当控制和确认。执行检查的操作员应至少每年接受目检资质确认(如果是佩戴眼镜的员工,在接受资质确认时也应佩戴矫正镜片)。确认应使用来自生产商缺陷库的适当样本组,并考虑最差条件(例如,检查时间、传送产品给操作员的传送带系统的线速度、容器尺寸或疲劳程度),并且应包括对视力检查的考虑。应尽可能避免操作员的注意力分散,并应在检查时经常进行适当的休息。 | ||
| Operation 操作 | During the 100 % inspection each object should be inspected for at least 5 seconds against a white background and an additional 5 seconds against a black background. Times may be shorter when using a semi-automatic system. The objects should be slightly twisted or slowly rotated to swirl up particles whilst avoiding the formation of air bubbles. 在 100% 检查期间,应在白色背景上检查每个物体至少 5 秒钟,并在黑色背景下额外检查 5 秒钟。使用半自动系统时时间可能会更短。应轻微扭曲或缓慢旋转物体以使颗粒旋转,同时避免形成气泡。 The post inspection recovery time for visual Inspection staff is of essential importance. The maximum time for continuous inspection activity between breaks and the total maximum inspection time for a shift/workday must be defined. 检查后的恢复时间对于目检人员来说至关重要。必须定义休息之间连续检查活动的最大时间以及一个班次/工作日的总最大检查时间。 Industry practice is 20-60 minutes of inspection. A current good practice goal is 30 minutes of inspection followed by a break of at least 5 minutes continued for a total maximum duration goal of no longer than 4 hours. All Maximum uninterrupted inspection activity has to be qualified. 行业惯例是20至60分钟的检查。当前良好的做法是检查 30 分钟,然后休息至少 5 分钟,总最长持续时间目标不超过 4 小时。所有最大不间断检查活动均须合格。 | Background and contrast: Contrast between the defect of interest and the surrounding background is required for detection. Increased contrast improves detection. The use of both black and white backgrounds is described in790, as well as other global pharmacopeias. Matte/nonglossy backgrounds are recommended to avoid interference from reflection. The use of both backgrounds provides good contrast for a wide range of particulate and container defects, which can be light or dark in appearance. Inspection rate: Enough time must be provided to allow for thorough inspection of each container; 790 specifies a reference time of 10 s/container (5 s each against both black and white backgrounds). Larger or more complex containers may require additional time for inspecting all attributes. Inspecting for extended periods of time can cause inspector fatigue and a decrease in inspection performance. On the basis of industry experience (46), it is recommended that inspectors be given a documented break from performing inspection at least every hour. This break should allow time to rest the eyes and mind and may be achieved with a short rest (e.g., 5 min) or a longer meal break. This need for regular breaks may also be met through rotation to a non-inspection function, such as material handling or documentation. |
| 对比解读: 1、ECA规定了黑背景至少观察5秒,白背景至少观察5秒;1790规定每个背景约5秒,对于较大或较复杂的容器可能需要更多的观察时间,同时1790指出延长的观察时间可以导致分析员疲劳,继而导致人员检测能力的下降。 2、ECA规定使用半自动系统时,检查时间可能相对较短。 3、ECA中规定单次最大观察时间的业界一般要求是20-60分钟,最佳实践建议观察30分钟,休息至少5分钟,目检的最长持续观察时间不超过4小时;USP建议最长观察时间为一小时,然后休息5分钟或者更长时间进餐等,定期休息也可以通过其它非目检的操作来实现,例如物料准备、记录书写等。 延伸补充观点: 中国药典:将供试品置遮光板边缘处,在明视距离(指供试品至人眼的清晰观测距离,通常为25cm),手持容器颈部,轻轻旋转和翻转容器(但应避免产生气泡),使药液中可能存在的可见异物悬浮,分别在黑色和白色背景下目视检查,重复检查,总检查时限为20s。 | ||
半自动化目检
| 要求 | ECA | USP 1790 |
| Semi-Automated Inspection 半自动化目检 | The complete inspection of the unit is ensured by: 通过以下方式确保对受检单元的全面检查: 1. To avoid disturbances of the inspector an inspection booth should exclude external light sources. 1. 为了避免对检查员造成干扰,目检间应屏蔽外部光源。 2. At least a full 360°rotation of the unit in the inspection field facilitates inspection. 2. 受检单元在目检视野区至少可以360°旋转,以方便检查。 3. Inspection of the top and the bottom of the unit ensures complete inspection of the unit. 3. 检查受检单元的顶部和底部,确保全面目检。 4. For liquid products particle inspection can be supported by pre-rotating the objects before inspection to get the particles moving within the solution. 4. 对于液体产品的颗粒物目检,在检查前旋转受检物品以使颗粒物在溶液内移动。 5. Due to the nature of a semi-automated inspection the inspection field is typically illuminated at around 8.000 lux or higher. Difficult to inspect objects, e.g. brown glass vials or colored solutions, may require illumination above 10.000 lux. 5. 由于半自动化目检的性质,目检视野区的照度通常在8,000 lux或更高。难以目检的物品,例如棕色玻璃瓶或有色溶液,可能需要10,000 lux以上的照明。 6. For an undisturbed visual inspection, the background of the booth should be black. Reflecting surfaces should be avoided. 6. 为了目检不受干扰,目检间的背景应为黑色。应避免光反射表面。 7. Magnification should be used in order to bring the visual focus point of the operator's eyes to the point of inspection using an ergonomic sitting situation of the operator. 7. 应使用放大功能,以便使操作员在采用符合人体工程学的坐姿时眼睛的视觉焦点能对准检查点。 Risk Management Principles via assessments should be used during qualification of all the variable parameters noted above. Pre-start up tests should be established and then implemented in a controlled manner to be used periodically. Functional control kits should be used to ensure variable parameters are within the validated state before commencement of the inspection process, whereas potential drifts of the equipment (e.g., Illumination) is more suited to be verified periodically, e.g. every six months. 在对上述所有可变量进行确认时,应进行评估并运用风险管理原则。应建立启动前测试,然后以受控的方式定期测试。应使用功能控制套件来确保在开始目检之前可变量处于验证状态,设备的潜在漂变(例如照明)更适合定期确认,例如每六个月一次。 The most important parameters are: 最重要的参数是: Conveyor Speed 传送带速度 Illumination 照度 pre-spinning / if applicable 预旋转/如适用 Rotation within the inspection field to ensure 360 degree inspection of the unit 在检查区域内旋转,确保对设备进行 360 度检查 Validation of the semi-automated inspection process must show at least equivalence to manual inspection results. 半自动化检查过程的验证必须至少与手动检查结果相当 For lyophilized products inspectors are qualified at maximum inspection speed and may inspect during routine operation at maximum or any lower inspection speed. 对于冻干产品,目检员以最大目检速度通过资质确认,常规操作中的目检速度可以≤最大目检速度。 For liquid products inspection the qualified speed must be routinely used at all times due to the influence of speed on the inspection validity. 对于液体产品,由于目检速度会影响目检的有效性,常规操作中的目检速度应始终为通过资质确认的目检速度。 After an inspection process of an uninterrupted and predefined duration, operators require an eye break. Semi-automated visual inspection requires greater concentration by the operator. For that, times of uninterrupted inspection are typically shorter and times of breaks may differ from the numbers given in chapter 2.3 (Operation of the Manual Visual Inspection). 经过一段预定时长的持续目检后,操作员需要让眼睛得到休息。半自动化目检需要操作员更加集中注意力。因此,持续目检时间通常较短,休息时间可能与第 2.3 章(人工目检操作)中的不同。 | These systems often use a conveyor equipped with rollers to transport the containers in front of the inspector inside an inspection booth or station. For inspection of liquids, the booth can be equipped with a high-speed spin station to set particles in motion. The rollers can also be used to slowly rotate the containers in front of the inspector as they traverse the inspection zone. These systems offer a means to control the presentation of the vials and can offer additional lighting options, such as Tyndall lighting, which may enhance the appearance of some defects such as cracks or small particles. Mirrors may also be used to provide a clear view of the top and bottom of each container. Rejected units may be removed from the rollers by hand, whereas some systems are equipped with a remote rejection system that can be triggered by the inspector. Care should be taken in the qualification and operation of these systems to ensure full rotation of vials in the inspection zone to assure examination of all surfaces. In addition, studies should be conducted to ensure the detection of heavy particles, which may not be lifted from the bottom of the container, and to ensure that the rate of inspection produces an acceptable detection rate for defects of interest CRITICAL PROCESS PARAMETERS FOR SEMI-AUTOMATED INSPECTION Light intensity must be controlled, as with MVI. The rate of inspection is controlled by the speed of the roller/conveyor or some equipment that allows the inspector to call for a group of containers each time. Spin speed for liquid products and rotation rate for all containers should be established during validation/qualification and maintained within the validated range for routine inspection. The background color is controlled by the color of the rollers selected and the color of the background seen through the spaces between the rollers. Qualification of inspectors and validation of the inspection equipment should be based on comparison with the compendial single-container manual-inspection process with an expectation that alternative methods such as semi-automated inspection demonstrate equivalent or better performance. |
| 对比解读: 1、ECA指南中对于半自动目检的要求更加具体和详细,更具有指导性。 2、ECA和1790 均要求半自动化目检程序应与人工目检等效或者更优。 3、半自动目检的关键控制参数:传送带速度、照度、预混合、360°观察等。 4、1790中指出半自动目检的背景颜色控制可以通过滚轴的颜色和滚轴间的空隙颜色控制。 5、ECA规定半自动化目检需要操作员高度集中注意力,为此,不间断检查的时间通常较短。 6、ECA规定,对于冻干产品,目检员以最大目检速度通过资质确认,常规操作中的目检速度可以≤最大目检速度 7、ECA规定,对于液体产品,由于目检速度会影响目检的有效性,常规操作中的目检速度应始终为通过资质确认的目检速度。 | ||
自动检查
| 要求 | ECA | USP 1790 |
| 4.1 Qualification / Validation | The central aspect of qualification/validation of a fully automated inspection system is the verification that the automated system is at least as good as the reference human inspector (without magnification) with regards to defect detection rates. Qualification and validation can be done consecutively or combined. The performance qualification of the system can be rated as validation of the process if it is carried out product-specific. In the qualification phases before the PQ, format-specific test sets can also be used. Bracketing concepts are possible. In this case product specific qualification sets have to be used. During machine qualification the inspection of a qualification set should be repeated at least 10 times. The detection rate should be compared to the results of a qualified manual inspection and should be at least as good as manual inspection based on defect categories. It is worth mentioning that an automated inspection machine is not capable of detecting and removing certain categories of cosmetic defects. 全自动检测系统鉴定/验证的核心方面是验证自动化系统在缺陷检测率方面至少与参考人工检查员(未放大)一样好。资格认定和验证可以连续进行,也可以合并进行。如果系统的性能鉴定是针对特定产品进行的,则可以将其评定为流程的验证。在 PQ 之前的资格认证阶段,也可以使用特定格式的测试集。括号概念是可能的。在这种情况下,必须使用特定于产品的资格集。在机器鉴定期间,鉴定组的检查应重复至少 10 次。检测率应与合格的人工检查结果进行比较,并且至少应与基于缺陷类别的人工检查一样好。值得一提的是,自动检测机无法检测和去除某些类别的外观缺陷。NOTE: The detection of particles follows a probabilistic detection principle depending on the size and potential physical movement of the particle in the drug product solution being inspected. The detection rate of particles needs to be viewed following the principle of the so called "Knapp-statistics". Julius Knapp demonstrated that a ratio of not more than 25% defect vials within a set of non-defect vials (good vials) gives the best statistical return for the accurate statistical evaluation of the particle detection rate. A man-machine comparison for introduction of a new or automated system of inspection is carried out by multiple (10X) detection rates of a test set of vials containing particles, (reject vials). Success is when results from the 10 repeats by the machine and the resulting test statistics are equally as good as when the 10 times repeated vial inspection is performed manually. This is the so-called Knapp-test. 注:颗粒的目检遵循概率检测原则,取决于被检药品溶液中颗粒的大小和可能的物理运动。需按照“Knapp 统计”原理估计颗粒的检出率。Julius Knapp 证明,一组无缺陷样品瓶(合格品)中混入缺陷样品瓶的比例不超过25%,颗粒检出率的统计学估计最准确。引入新目检系统或自动化目检系统时,准备一个测试集(其中包含含颗粒的样品瓶(不合格品)),进行人工目检-机器目检的对比,检出率放大10倍(10X)。当重复10次机器目检的结果和由此产生的测试统计值与重复10 次人工目检的的结果和统计值一样好时,即为成功。这就是所谓的 Knapp 测试。 | AVI offers advantages in the areas of throughput and consistency, compared with MVI (5). AVI may also offer enhanced sensitivity for some defects, compared with MVI, but may suffer from higher false rejection rates due to the inability to tolerate normal variation in containers or product. This is especially true for molded glass containers and flexible bags. Validation of the automated inspection equipment should be based on comparison with the compendial manual inspection process with an expectation that alternative inspection methods demonstrate equivalent or better performance. Significant effort is required to program these systems and to test their performance against a range of known defects, as well as acceptable containers. ➣ LIGHT OBSCURATION METHODS ➣ IMAGING METHODS |
| 4.2 Routine Operation 日常操作 | During routine operation the overall functional performance of the automated system should be demonstrated to be within the acceptable range of the normal operating conditions and validated state. This is achieved by using a test set containing representations of the range of defects that has been used in the qualification, which should be applied before and after the inspection of a batch. An abridged or reduced function set may also be used. In the event of a risk, such as a malfunction or machine drifts (misalignments of visual components, camera defects, etc.) of the inspection system, the described functional test can also be useful within a batch 在常规运行期间,应证明自动化系统的整体功能性能在正常运行条件和验证状态的可接受范围内。这是通过使用包含鉴定中使用的缺陷范围表示的测试集来实现的,该测试集应在批次检查之前和之后应用。还可以使用简化或缩减的功能集。如果出现风险,例如检查系统发生故障或机器漂移(视觉组件错位、相机缺陷等),则所述功能测试在批次内也很有用 | |
| 4.3 Requalification | Requalification of an automated inspection system should be ideally carried out annually, or every two years at the latest. This must be done by evaluation of the changes and deviations that occurred during the period of operation. This review must include a statistical trend analysis of the performance data obtained during routine inspection and system suitability determinations using the function sets before and after every machine use. 自动检测系统的重再确认最好每年进行一次,或者最迟每两年进行一次。这必须通过评估运行期间发生的变化和偏差来实现。该审查必须包括对例行检查中获得的性能数据的统计趋势分析以及每次使用机器之前和之后使用功能集进行的系统适用性确定。 | |
| 4.4 Revalidation | An automated visual inspection machine must be considered a critical system. Therefore, a periodic revalidation of the operation should be carried out, e.g. ever 3-5 years. Revalidation (as any validation) is product specific. Bracketing approaches for the revalidation are possible. 自动视觉检查机必须被视为一个关键系统。因此,应定期对操作进行重新验证,例如每3至5年一次。重新验证(与任何验证一样)是特定于产品的。可以采用括号方法进行重新验证。 Different approaches may be used: 可以采用不同的方法: Revalidation can be done by repeating the verification of the human inspection versus the inspection results of the machine, for example by manual re-inspection of an automatically inspected batch or part of the batch (e.g. 5000 vials). The acceptance criteria are the same as during the initial validation. 可以通过重复验证人工检查与机器检查结果来进行重新验证,例如通过手动重新检查自动检查的批次或部分批次(例如 5000 瓶)。验收标准与初始验证时相同。 Revalidation may also be done by a continuous revalidation approach using the AQL results. The AQL is a manual inspection of a representative sample performed for every inspected batch (man-machine comparison), constituting a repeated verification of human inspection versus the automated inspection machine on batch by batch level and thus be considered a revalidation. 还可以使用 AQL 结果通过连续重新验证方法进行重新验证。AQL 是针对每个检验批次进行的代表性样本的人工检验(人机比较),构成了人工检验与自动检验机器逐批重复验证,因此被视为重新验证。 Result and evaluation should be documented in a revalidation report. 结果和评估应记录在重新验证报告中。 | |
| 对比解读: 1、ECA和1790均要求自动检查需要和人工目检进行对比,证明其等效或者更优,ECA规定了具体的测试方法和标准Knapp测试,同时1790指出了自动目检可能会有更高的误检率。 2、1790列举了自动检查的两种技术,光阻法和照相法,并描述了方法的适用范围。 3、ECA规定了自动目检机日常确认的要求,测试前后。 4、ECA规定了自动目检机定期回顾的要求,每年或最多两年一次。 5、ECA规定了自动目检机再验证的要求,建议3-5年一次。 | ||
冻干产品目检
| 要求 | ECA | USP 1790 |
| Inspection of lyophilized product 冻干产品目检 | Lyophilized drug products also have to undergo a 100% visual inspection. However, as particles can be detected only on the surface of the product cake and not in the cake itself, an additional test on particulate matter has to be performed. For this, a number of samples are reconstituted and visually inspected for particulates. The result has to be within the predefined particle limits. As this supplemental testing is destructive in nature, it is acceptable that only a limited number of samples are inspected. Samples from the batch are taken according to a sampling plan (e.g. S-3 and/or S-4 according to DIN ISO 2859). 冻干药品也必须经过100%目检。然而,由于只能检出冻干粉饼表面的颗粒物,而不能检出冻干粉饼本身存在的颗粒物,因此必须进行额外的不溶性微粒检查。为此,复溶一定量的样品,并目检复溶溶液中是否含有颗粒物。结果应在预先界定的粒子限值范围内。由于该补充检查本质上具有破坏性,因此只有在仅检查有限数量的样品的情况下才能接受进行该检查。根据取样计划从批次中抽取样品(例如根据 DIN ISO 2859 的 S-3 和/或 S-4)。 | Typical sampling plans for supplemental testing can be found in the special sampling plans S-3 and S-4 in ANSI/ASQ Z1.4 (42). These higher sensitivity S-plans offer a practical compromise between sample size and statistical power. For batch sizes between 200 and 100,000 they suggest a sample size of 20 with an accept number of 0 (based on an AQL of 0.65%). Sample sizes larger than 20, as found in these sampling plans, may be appropriate for larger batch sizes or when additional sensitivity to support reduced AQL values or a more conservative defect classification is desired. |
| 对比解读: 1、由于冻干产品的特殊性,ECA和1790均规定了冻干产品100%目检后需要进行复溶后的补充测试,取样计划建议为ISO 2859 的 S-3 和/或 S-4,1790中对于批量200-100000建议的取样量为20支(基于AQL 0.65%)。 2、1790除冻干产品外,对于较难目检的产品(剂型、包装及给药系统等)列出了推荐的补充测试建议,例如:粉针产品、乳剂、混悬剂、细胞及基因治疗制剂,茶色/琥珀色玻璃、半透明塑料包装产品,大容量制剂,组合产品等。 3、1790中规定,对于包装类型限定目视检查时,可以采取替代的补充测试方法,例如转移、过滤、离心、过筛等。 延伸补充观点: 对于冻干产品,需要注意的是在复溶过程中,应在洁净的环境中进行,避免造成假阳性的情况,更多可以参考PDA TR79 Particulate Matter Control in Difficult to Inspect Parenterals 2018 | ||
缺陷品分类
| 要求 | ECA | USP 1790 |
| Defect Classes | There should be at least two product-specific defect classes defined. Defining more defect classes may be appropriate, e.g.: 应该至少定义两类特定于产品的缺陷。定义更多的缺陷类别可能是合适的,例如: Critical defects: may cause a lack of sterility, container integrity or cause harm to patients 关键缺陷:可能导致无菌性问题、容器完整性问题或导致患者伤害 Major defects: may alter the content or the function of the product 主要缺陷:可能会影响产品的内容物或功能 Further defect classes can be: 更多缺陷类别可以是: Minor defects: Defects that do not affect patient health or product functionality 次要缺陷:不影响患者健康或产品功能性的缺陷 If a company performs additional visual inspection for specific cosmetic appearance defects for products intended to be placed to culturally sensitive markets it is recommended not to include them into the GMP defect classification. 对于拟投放到文化敏感市场的产品,如果企业进行特定外观缺陷的额外目检,建议不要将其纳入GMP缺陷类别。 | Defects are commonly grouped into classifications based on patient and compliance risk (2). The most common system uses three groups: critical, major, and minor. Critical defects are those that may cause serious adverse reactions or death of the patient if the product is used. This classification includes any nonconformity that compromises the integrity of the container and thereby risks microbiological contamination of the sterile product. It may also include anomalous extrinsic particle types such as insects or other filth or adulteration. Major defects carry the risk of temporary impairment or medically reversible reactions or involve a remote probability of a serious adverse reaction. Visible intrinsic particulate matter typically falls into the major defect category. This classification is also assigned to any defect that impairs or makes the product unusable. Minor defects do not impact product performance or compliance; they are often cosmetic in nature, affecting only product appearance or pharmaceutical elegance |
| 对比解读: 1、对于缺陷品分类,ECA和1790的规定一致,建议按照关键缺陷、主要缺陷和微小缺陷,从缺陷对于产品质量和病人用药风险角度来分级。 2、对于拟投放到文化敏感市场的产品,如果企业进行特定外观缺陷的额外目检,ECA建议不要将其纳入GMP缺陷类别。 3、1790 详细列出了异物标准品、异物标准品制作及测试集等相关要求。 延伸补充观点: 1、对于新建车间、新的产品品种或者剂型等,在初期因为生产批次少,不能收集足够的缺陷样品,或者有些缺陷类型很少发生,所以不得不自己制备或者委外制备。如果是自己制备,那么首先必须了解产品的特性,以及可能存在的缺陷类型有哪些,比如产品里有纤维、胶塞碎片、玻璃碎片等等。那么就需要选择一些合适的材料进行模拟,不仅仅要考虑这些颗粒,还需要考虑产品的特性,如是悬浊液还是透明液体还是冻干产品,容器的装量等等,都尽可能的与实际产品保持一致,才能具有代表性。具体的做法可以文献Considerations for Design and Use of Container Challenge Sets for Qualification and Validation of Visible Particulate Inspection,国内可以参考JB/T 20135安瓿注射液异物自动检查机的附录A 试瓶制作部分。 2、BPOG: BioPhorum Operations Group Ltd在2023年1月发布《100%目检并不意味着100%缺陷检测》,旨在纠正一个普遍的误解:即认为对生物制药产品的100%目检等同于100%的缺陷检测。尽管产品经过了全面的目检,但由于目检过程本身的概率性质,并不能保证能够系统性地识别所有缺陷。目检缺陷的关键性和可检测性,这两者并不直接相关,因此即使进行了100%的目检,也不能保证100%的缺陷被检测出来。因此100%的目检并不等同于100%的缺陷检测,而应该被视为一个概率性过程,并且需要结合风险管理和质量控制策略来优化目检过程。 | ||
缺陷品评估及趋势分析
| 要求 | ECA | USP 1790 |
| 7.1 Manual, semi-automated and fully automated (non-inline) inspection | Based on the trend analysis of the production process a limit for each defect class should be defined. There should be limits for individual defects and for the sum of all defects within a defect class. Yields should also be monitored. 应根据生产过程的趋势分析来定义每种缺陷类别的极限。对于单个缺陷以及缺陷类别中所有缺陷的总和都应设有限制。还应监测产量。 Limits should be defined on process history (overall maximum reject rate, rate per defect / particle category) and should reflect and reference the process capability index (CpK) for the process step. 应根据工艺历史(总体最大拒收率、每个缺陷/颗粒类别的拒收率)定义限值,并应反映和参考工艺步骤的工艺能力指数 (CpK)。 Typical action limits for individual defects are for example: 个别缺陷的典型行动限制例如: Critical defects: 0.5 % to 1 % * 严重缺陷:0.5% 至 1% * Major defects: 1 % to 3 % 主要缺陷:1% 至 3% Minor defects 3 % to 5 % 微小缺陷 3% 至 5% *There are critical defects (e.g. turbidity) with an acceptance limit of 0. *存在严重缺陷(例如浑浊度),接受限度为 0。 Typical action limits can also be set on specific defects (e.g. particles) instead of using the categories critical, major and minor. 还可以针对特定缺陷(例如颗粒)设置典型的操作限制,而不是使用严重、主要和微小类别。 Setting alert limits may be reasonable e.g. to identify a production specific problem as early as possible. 设置警报限值可能是合理的,例如,以便尽早发现特定生产问题。 | Data obtained from the in-process 100% inspection followed by AQL inspection are used for batch release. Both 100% and AQL inspection data should also be analyzed for adverse trends on a periodic basis, typically at least annually. Data from the 100% inspection provides the best source of typical defect types and rates during normal production. High-volume products may generate enough data to allow quarterly analysis, whereas a longer period may be necessary to accumulate data for products that are produced infrequently. Data from component inspection, production 100% inspection, and the AQL inspections should be evaluated based upon sound statistical principles to determine whether the current action levels accurately reflect current process capability. Alert and/or action levels may be established and/or adjusted if the statistical analyses indicate that lower defect levels are being observed consistently. When establishing new action or alert levels, a preliminary target value may be used until enough production experience is obtained. Consideration should be given to planned improvements in the manufacturing and inspection processes. If significant improvements are planned, the reduction of the action/alert level should not be instituted until the impact of the improvement is measured over enough time to establish the validity of the new value. Reinsertion should only be conducted using a procedure that has been approved by the quality organization and addresses key parameters such as the inspection conditions (e.g., same as primary inspection or modified to enhance detection of a specific defect type), the number of times reinspection may be performed (this should be limited and justified), and the acceptance criteria (e.g., same as primary inspection or tightened). If reinspection is required often, consideration should be given to improving the sensitivity of the primary inspection process or of the manufacturing controls to prevent defects in the upstream process as determined by root cause analysis. |
| According to ASTM E2587-2 (Standard Practice for Use of Control Charts in Statistical Process Control) control limits for trending should be calculated from process data and not be based on fixed specification limits. However, the evaluation should optimally be based on 30 representative data points of the process. This would require preliminary control limits to be defined based on experience prior to the first calculation based on measured data points. 根据 ASTM E2587-2(统计过程控制中控制图使用的标准实践),趋势的控制限应根据过程数据计算,而不是基于固定的规格限。然而,评估最好应该基于该过程的 30 个代表性数据点。这需要在基于测量数据点进行第一次计算之前,根据经验定义初步控制限值。 The measures to be taken for batches exceeding these limits are to be predefined. The re-inspection of a batch due to a limit violation should be investigated. In certain circumstances the method of inspection used in the initial inspection may not be suitable for the reinspection depending on defect detection rate. The decision on the reinspection method has to be made by a suitable authorised and qualified individual. 对于超出这些限制的批次要采取的措施是预先定义的。应对因违反限制而进行的批次重新检验进行调查。在某些情况下,根据缺陷检测率,初次检查所使用的检查方法可能不适合重新检查。重新检查方法的决定必须由具有适当授权和资格的人员做出。 | ||
| This assessment should be part of the investigation. For example: batches exceeding one time the acceptance limit should trigger a failure investigation. Batches exceeding the acceptance limit two times should be additionally 100 % re-inspected. Re-inspection should never be performed without a previous investigation why limits have been exceeded. Re-inspection should be performed independent of the initial technique used - manual, semi-automatic or automatic visual inspection, but should be limited to not more than 2 re-inspections. Limitations of the inspection system should also be taken into consideration. If an automated system fails in identifying certain defects in the first run, it is likely to fail also in a second run. 该评估应成为调查的一部分。例如:超过验收限度一倍的批次应引发失败调查。对于两次超出可接受限度的批次,还应进行100%的复检。如果没有事先调查过超出限制的原因,就不应该进行重新检查。重新检验应独立于最初使用的技术(手动、半自动或自动目检)进行,但重新检验次数应限制为不超过 2 次。还应考虑检查系统的局限性。如果自动化系统在第一次运行中无法识别某些缺陷,则很可能在第二次运行中也会失败。 Re-inspection of rejected containers is not recommended and must not be performed without justification based on a thorough investigation. 不建议对被拒收的容器进行重新检验,并且不得在没有经过彻底调查的合理依据的情况下进行重新检验。 | ||
| "Grey Channel" for fully automated inspection systems: Defining a "grey eject channel" in automated inspection may be useful to separate containers for which the inspection result is not clear. From a technical point of view, a "grey channel" is also meaningful in case of e.g. machine stops, when it is uncertain whether a vial has been fully inspected or not. 全自动检测系统的“灰色通道”:在自动化检测中定义“灰色弹出通道”可能有助于分离检测结果不明确的容器。从技术角度来看,“灰色通道”在机器停止等情况下也是有意义的,此时不确定小瓶是否已经完全检查。 Examples for the Grey Channel: 灰色通道的示例: 1. Air bubbles: Air-bubbles might be the reason for an unclear inspection result. Camera systems cannot distinguish between particulate matter and air-bubbles. Therefore, re-inspection of containers from the grey channel after a certain holding time of the product vials in order to reduce the air-bubbles is allowed but should not be performed more than once. 1. 气泡:气泡可能是导致检查结果不清晰的原因。摄像系统无法区分颗粒物和气泡。因此,为了减少气泡,允许在产品瓶保持一定时间后从灰色通道重新检查容器,但不应进行多次。 2. Other defects in the grey channel: From a technical point of view some small defects (e.g. small scratches) should be re-inspected manually if the camera system is not capable of detecting the defect. The feasibility is shown during the validation. 2. 灰色通道中的其他缺陷:从技术角度来看,如果摄像系统无法检测到缺陷,则应手动重新检查一些小缺陷(例如小划痕)。验证过程中显示了可行性。 Objects in the grey channel should only be re-inspected one time. Trending is done over the whole batch after the objects have been classified as good or defects. 灰色通道中的物体仅应重新检查一次。在将物体分类为良好或有缺陷后,对整个批次进行趋势分析。 | ||
| 7.2 Automated inspection: In-line | An in-line fully automated inspection system should also trend and analyse the production process. Due to the in-line process and the fact that camera systems cannot trend defect categories a trending system has to be established on the technical level e.g. side-wall defect, crimping-defect, shoulder defect etc. Limits of these cameras and their inspection scope have to be established according the ASTM E2587-2 standard. This gives a reference to the process capability index (CpK) for the in-line controlled process. 在线全自动检测系统还应对生产过程进行趋势分析。由于在线流程以及摄像系统无法趋势化缺陷类别的事实,必须在技术层面建立趋势系统,例如侧壁缺陷、卷曲缺陷、肩部缺陷等。这些摄像机的限制及其检查范围必须根据 ASTM E2587-2 标准确定。这为在线控制过程的过程能力指数(CpK)提供了参考。 The measures to be taken during the batch production exceeding these limits need to be predefined and should lead to actions during the batch production process. Herein a re-inspection of the batch is not needed due to the immediate corrective action. 超过这些限制的批量生产期间需要采取的措施需要预先定义,并且应该在批量生产过程中采取行动。由于立即采取了纠正措施,因此无需对该批次进行重新检验。 Re-inspection of rejected containers is not recommended and must not be performed without judgment done within in an investigation. 不建议对被拒收的集装箱进行重新检验,且未经调查判断不得进行重新检验。 | |
| 对比解读: 1、ECA和1790均要求基于历史数据进行缺陷趋势分析,建立警戒线和行动线。 2、ECA建议趋势分析应反映和参考工艺步骤的工艺能力指数 (CpK),并给出了建议的行动线参考值和趋势分析的方法。 3、ECA中建议在自动化检测中定义“灰色弹出通道”,可能有助于分离检测结果不明确的容器。 4、对于重复目检,ECA和1790均要求限制,不超过2次,重复目检需要论述和批准后执行。 | ||
批次放行
| 要求 | ECA | USP 1790 |
| BATCH RELEASE 批放行 | For the release decision two criteria need to be evaluated: 对于放行决策,需要评估两个标准:
The results of the 100% visual inspection, done as part of the manufacturing process, are an integral part of the batch documentation, specifying the types of defects found (fibre, turbidity, crack, etc.) as well as a classification of the defect such as critical, major or minor. Acceptance criteria must be pre-defined for these defect classes of all defects found during the 100% inspection process (see above). For the automated in-line inspection these classifications needs to be done on a technical level (side-wall defect, crimping defect,…) and cannot be done using the classification such as critical, major or minor (see above). 100%目检是生产过程的一部分,其结果是批文件记录的组成部分,其中指定了发现的缺陷的类型(纤维、浑浊、裂缝等)以及缺陷的类别,例如严重、主要或次要缺陷。对于100%目检中发现的所有缺陷,必须按照缺陷类别预先界定可接受标准(见上文)。对于自动在线目检,不能使用诸如关键缺陷、主要缺陷或次要缺陷的缺陷类别,而需从技术层面(侧壁缺陷、轧盖缺陷等)进行分类(见上文)。 For the AQL manual inspection a randomized sampling of the 100% inspected batch should be performed according to a pre-determined AQL procedure. AQL manual inspection can be carried out by production staff under a quality oversight or the quality unit. 对于 AQL人工目检,应根据预先确定的AQL 程序对100%目检批次进行随机抽样。AQL人工目检可由生产人员在质量监督下进行或由质量部门进行。 | During 100% inspection, limits on typical rejection rates should be established to identify atypical lots (40). These limits may be established for categories of defects (e.g., critical, major, and minor) or for specific types of defects (e.g., particles). A review of historical performance is useful in establishing these limits, and the review may include grouping products similar in appearance and manufacture. Periodic reassessment of these limits is recommended to account for expected process improvements and/or normal fluctuations in the process baseline (41). If a limit is exceeded, it should trigger an investigation. The investigation may include an additional inspection, or it may determine whether additional inspection is necessary. After 100% inspection, a statistically valid sample is taken from the units accepted by the inspection process. These sampled units should be manually inspected under controlled conditions by trained inspectors. Chapter α790ρ provides reference inspection conditions for this purpose. The sample may be a random or a representative sample (e.g., at fixed time intervals or a fixed number per tray). Defects may not be distributed equally over the lot, and therefore a sampling process that represents the whole lot is required. Typical sampling plans used for this purpose can be found in ANSI/ASQ Z1.4 (42). Equivalent plans may also be found in ISO 2859 (43) or JIS Z9015 (44). These plans were historically developed for larger lot sizes and thus oversampled smaller batches (45). Alternative sampling plans may be justified for smaller batch sizes resulting in a smaller sample size. For batch release, the sampling plans listed as Normal II are typically used. Tightened sampling plans may be appropriate when an atypical result is observed, or reinspection is performed. If the acceptance criteria of the sampling plan are not met, an investigation should be conducted. Depending on the nature of the failure, this investigation should include forensic classification/identification of the particle(s), and examinations of the manufacturing process, the raw materials, and the packaging materials, as well as the inspection process. If, after investigation, the inspection process is deemed capable of detecting the defect(s) in question, the batch may be reinspected. An alternative inspection process better suited to detection of a specific defect may also be chosen for reinspection. After reinspection (performing a second 100% inspection of the batch), a new statistically valid sample of the accepted units is taken and compared against established acceptance criteria. It is a good practice to use a tightened sampling plan and acceptance criteria under these circumstances because of the atypical nature of this process step. |
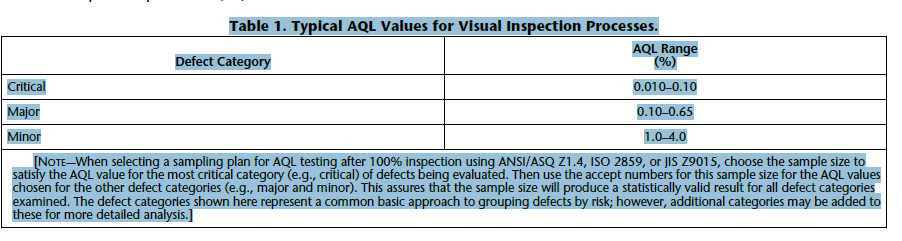
| AQL | For release of the batch, the minimum AQL should be为了放行批次,最低 AQL 应为 Critical: 0 defects within the AQL samples 严重:AQL 样品中 0 个缺陷 Major: ≤ 0.65 AQL 主要:≤ 0.65 合格标准 Minor: ≤ 4.0 AQL 微小:≤ 4.0 合格质量限 If an AQL limit is exceeded, the whole batch may be re-inspected 100% followed by a second AQL manual inspection (see 6.). This process can be repeated, but must be justified by an investigation and should not be performed more than 2 times in maximum. It is recommended to have tighter AQL limits for the repeated AQL testing. Tightening of AQL limits is possible by two ways: Stay on the AQL sample size and use the next (tighter) AQL limit- OR: Stay on the AQL limit and use the next (higher) AQL sample size. 如果超出 AQL 限制,则可能要对整个批次进行 100% 重新检验,然后再进行第二次 AQL 人工检验(参见 6.)。该过程可以重复,但必须通过调查证明其合理性,并且最多不应执行超过 2 次。建议对重复的 AQL 测试设置更严格的 AQL 限制。可以通过两种方式来收紧 AQL 限制:保留 AQL 样本量并使用下一个(更严格的)AQL 限制 - 或者:保持在 AQL 限制并使用下一个(更高)AQL 样本大小。 | Critical defects (those that pose the greatest risk to the patient) should be assigned an AQL with a very low value. Often, the accept number (the number of defective units allowed in the sample) for a critical defect is zero. Major and minor defects, which pose less risk to the patient, will have increasing (less stringent) AQL values and accept numbers greater than zero. Further definition and discussion on defect categories are found in 5.1 Defect Classification. Table 1 shows the range of AQL values typically used for visual inspection processes (46). |
| AQL | According the USP particles are allowed to be within the AQL manual inspection. This is connected with AQL limits for particulates greater than 0.01 knowing that the AQL manual inspection is a "statistical tool" where a 100% assurance of having no visible particles within the batch is never given. Unfortunately, within the USP no statement regarding the criticality of particles is given. With this one should investigate the particle(s) found. Any particulate defects found in the AQL manual inspection must be evaluated to determine whether the particulate is characteristic of the filling process. This evaluation should include the nature, the size and the detection rate of the particle within the given Drug Product. If the particulate defect is not characteristic of the filling process, failure of the AQL may result. If the particulate defect is at the border of visibility, having a low and local defined detection rate, a re- inspection of the affected batch may not be necessary. 根据 USP,颗粒允许在 AQL 手动检查范围内。这与颗粒物 AQL 限值大于 0.01 有关,但 AQL 手动检查是一种“统计工具”,无法 100% 保证批次内没有可见颗粒。不幸的是,USP 中没有给出有关粒子临界性的声明。人们应该用它来调查发现的粒子。必须评估 AQL 手动检查中发现的任何颗粒缺陷,以确定颗粒是否为灌装过程的特征。该评估应包括给定药品内的颗粒的性质、大小和检测率。如果颗粒缺陷不是填充过程的特征,则可能导致 AQL 失败。如果颗粒缺陷位于可见性的边界,且检测率较低且为局部定义,则可能不需要对受影响的批次进行重新检验。 The number of AQL manual inspection steps should be evident in the batch documentation. 批次文件中应明确注明 AQL 手动检查步骤的数量。 | USP 790 Sampling at Batch Release (After 100% Manufacturing Inspection) Sample and inspect the batch using ANSI/ASQ Z1.4 (or ISO 2859-1). General Inspection Level II, single sampling plans for normal inspection with an AQL of 0.65%. Alternative sampling plans with equivalent or better protection are acceptable. NMT the specified number of units contains visible particulates. |
| 对比: 1、对于放行决策,ECA和1790均需要评估两个标准: 100%批目检的趋势分析。 AQL人工目检的结果。 2、对于建议的AQL最低标准,ECA和1790基本一致,均要求严重:AQL 样品中 0 个缺陷 Major: ≤ 0.65 AQL 主要:≤ 0.65 合格标准 Minor: ≤ 4.0 AQL 微小:≤ 4.0。 3、批放行的取样方案,1790建议选择一般抽样水平II。 4、对于超出 AQL 限制,ECA和1790规定一致,调查后可能要对整个批次进行 100% 重新检验,然后再进行第二次 AQL 人工检验,该过程可以重复,但必须通过调查证明其合理性,并且最多不应执行超过 2 次,建议对重复的 AQL 测试设置更严格的 AQL 限制。 5、对于可指定原因的高误检率(合格品误检为不合格品)的情况,例如气泡、特殊包装或胶塞差异,1790允许二步目检法,此种情况多对适用于自动目检机,对于这两步的检测,均需再分别进行AQL测试,结合两步测试的结果重新计算异物的POD。 6、ECA指出790中规定异物的检出应满足AQL的标准,建议的AQL为0.65,此AQL标准未基于异物的关键性,如果异物属于严重缺陷,则AQL应失败,因此,如果发现存在颗粒物,应进行调查和评估,该评估应包括异物的性质、大小和检测率。 | ||
已分销产品问题
| 要求 | ECA | USP 1790 |
| Concerns regarding distributed product 已分销产品问题 | When concerns regarding particulate matter of product already distributed to the market arise, it is recommended to inspect a number of samples following a practical DIN ISO 2859 compliant sampling plan. Visual inspection for particulates is a probabilistic process, therefore non-critical defect sampling plans with a limit of zero are not practical as an assessment tool and should be avoided. The inspection needs to be performed under the same conditions the routine inspection was performed. The relative batch and sample size will determine the acceptance criteria, if these criteria are met by inspection of these samples, the batch can be considered essentially free of particles. It is good practice to isolate the particle(s) of the complaint sample and to determine the size and to characterize the nature of the particle(s) within the investigation. 当对已分销到市场的产品的颗粒物产生担忧时,建议按照符合 DIN ISO 2859 的实际抽样计划检查一定数量的样品。颗粒物的目检是一个概率过程,因此,极限为零的非严重缺陷抽样计划作为评估工具并不实用,应避免使用。检查需要在与例行检查相同的条件下进行。相对批次和样本大小将决定验收标准,如果通过检查这些样本满足这些标准,则可以认为该批次基本上不含颗粒。优良作法是分离投诉样本的颗粒,并确定其大小并在调查中描述颗粒的性质。 | Chapter 790 states, "If it becomes necessary to evaluate product that has been shipped to customers (e.g., because of a complaint or regulatory concern), sample and inspect 20 units. If no particles are observed in the sample, the batch is considered essentially free of visible particulates. If available, additional units may be inspected to gain further information on the risk of particulates in the batch."Testing outlined in Product in Distribution is permissible only if Sampling at Batch Release (after 100% Manufacturing Inspection) has been successfully completed. Upon receipt, suspect containers should be subjected to the same inspection conditions and methodology used in the release inspection. Particle(s) verified in the returned or re-evaluated supply must be carefully characterized by an analytical forensic process to determine their source and likely cause. Single particles of typical product-contact materials are unlikely to present a concern. Multiple particles, large particle sizes, and any particles indicative of physical or chemical change are significant events and should be subject to further investigation. Rare instances of particulate material falling into the gray zone should be expected given the probabilistic nature of the inspection process and should not routinely trigger further evaluation of retention samples. The particle detection threshold should be determined for a specific inspection method and product/package combination incorporating a variety of particle types and densities that are typically found in the manufacturing environment. For example, the detection threshold for routine, reliable detection (≥70% probability) of a single spherical particle in a clear solution contained in a 10-mL vial utilizing diffuse illumination between 2000 and 3000 lux is often near 150 μm in diameter (5). Units returned from distribution may be false positive, may contain particle(s) larger than the acceptance threshold that were missed, may contain particle(s) in the gray zone (i.e., less than the detection threshold), or may have suffered a physicochemical change that resulted in a visible change. Ideally, there will be no visible particles in the containers released to market; however, there is always a low probability that this may occur. |
| 对比解读: 1、当对已分销到市场的产品的颗粒物产生担忧时,USP 790 建议的取样量为20支,如果未检测到异物,则认为基本无可见异物,如果检测到异物,需要测试更多的样品来收集相关的信息来判定异物的风险,需要注意的是,该评估仅适用于放行测试(100%目检后)成功完成并符合标准的情况。 2、对已分销到市场的产品的颗粒物产生担忧时,ECA建议按照符合 DIN ISO 2859 的实际抽样计划检查一定数量的样品,对于非关键缺陷,标准为零,并不实用,应避免使用。 3、对于异物的投诉,ECA和1790 均建议比较好的作法是分离投诉样本的颗粒,并确定其大小并在调查中描述颗粒的性质,基于异物的大小及种类进行评估。 4、该部分中的内容中,1790 提出了对于特定产品的目检检测方法需要进行不同种类异物检测限的研究。 | ||
总结分析
1、ECA 注射剂的目检4.0版本于2024.3发布,该指南视为对不同药典个论的额外补充,章节中对于半自动化检查、全自动化检查、缺陷品趋势分析的描述较1790更加具体详细和指导性。
2、1790对于人员资质确认的方法和可接受标准以及测试集的开发和制作的规定更加具体和具有指导性。
3、1790中对于注射剂工艺控制的要求更加详细具体,基于产品生命周期的角度综合去考虑,颗粒按照外源的、内在的或固有的进行划分,良好的产品开发和产品稳定性能够降低颗粒形成风险。
4、对于独特产品及特殊包装产品的目检可参考PDA TR79 Particulate Matter Control in Difficult to Inspect Parenterals 2018的相关规定。